A long-held belief is that testosterone stimulates development of prostate cancer and/or accelerates its growth. This fear is the most common reason for doctors’ reluctance to prescribe testosterone replacement therapy, even in hypogonadal men [1, 2] , which unnecessarily deprives many hypogonadal men of clinical benefits.
This summary gives an overview of an in-depth review of current literature regarding the relationship of testosterone levels and prostate cancer, and the effect of testosterone replacement therapy on prostate cancer progression and recurrence.[3] Key studies which have refuted the old belief that testosterone has harmful effects on the prostate are presented, along the new testosterone-prostate paradigm known as the saturation model.
Surprisingly, new research provocatively suggests that it is not high testosterone levels that are problematic for prostate cancer, but to the contrary that it is low testosterone that is associated with worrisome cancer features and outcomes…and new experimental research has uncovered mechanisms that explain how low testosterone levels may be detrimental for prostate health, and support the new view that testosterone therapy actually may have beneficial effects with regard to prostate cancer…
Key Points
* The longstanding concerns regarding putative detrimental effects of testosterone and testosterone therapy on prostate cancer, known as the “androgen hypothesis“, stem from several observations;[3]
– dependence of the prostate on androgens for normal development and function;
– beneficial response to androgen-deprivation therapy (ADT) in men with advanced or metastatic prostate cancer;
– historical reports of rapid prostate cancer progression in men who received testosterone administration;
– dramatic prostate specific antigen (PSA) declines in men with prostate cancer who undergo ADT;
– reduction in PSA and prostate volume in men with benign prostatic hyperplasia treated with 5alpha-reductase (5-AR) inhibitors;
– parallel rise in PSA and serum testosterone upon cessation of luteinizing hormone-releasing hormone (LH-RH) agonists (medications that lower the endogenous production of testosterone).
* The androgen hypothesis arose from two small studies in the 1940s in which men with metastatic Prostate cancer demonstrated clinical and biochemical improvement with androgen deprivation via castration, and rapid Prostate cancer progression with testosterone administration.[4, 5] However, these observations were made in a special population (castrated men) and are therefore not relevant to TRT in hypogonadal men.[6]
* The “saturation model” explains the paradoxical observations that prostate tissue is exquisitely sensitive to changes in serum testosterone at low concentrations but becomes indifferent to changes at higher testosterone concentrations.
* A threshold effect occurs in which increasing androgen concentrations reach a limit (the saturation point) beyond which there is no further ability to induce androgen-driven changes in prostate tissue growth.
* A mechanism contributing to the saturation model is the finite ability of androgens to bind to the androgen receptor (AR).
Maximal androgen–AR binding (i.e., saturation) occurs at fairly low androgen levels. It has been established in clinical practice that the saturation point appears to be around 230 ng/dL (8 nmol/L), subject to inter-individual variation.
* The saturation model explains the following important clinical observations:
– A comprehensive analysis of pooled worldwide epidemiologic data from 18 prospective studies, comprising a study population of 3886 men with prostate cancer and 6438 age-matched controls, found no relationship between prostate cancer risk and serum concentrations of testosterone, calculated free testosterone or DHT.[7]
– Two meta-analyses of testosterone therapy intervention studies, which specifically focused on analyzing potential adverse effects of testosterone therapy, did not find any significant differences in prostate outcomes between testosterone therapy vs. placebo treated men.[8, 9]
– In the UK Androgen Study, 1365 men 28–87 yr of age (mean 55) received testosterone therapy for up to 20 yr, with PSA and digital rectal examination (DRE) performed every 6month.[10] 14 new cases of prostate cancer, all localized, were detected after 1–12 years (mean 6.3 years). However, this prostate cancer is the same as in a general population which has never been treated with testosterone. Initiating testosterone treatment had no statistically significant effect on total PSA, free PSA or free/total PSA ratio, and any initial PSA change had no predictive relationship to subsequent diagnosis of cancer.[10]
– In a study on transdermal testosterone patch use for up to 6 years, PSA increased at 3 months from 0.47 to 0.60 ng/ml, followed by negligible change in PSA (0.03 ng/ml per year) over the remaining 5 yr. No Prostate cancer was identified in this trial.[11]
* The current ISA, ISSAM, EAU, EAA, ASA guidelines state:[12]
– There is no conclusive evidence that testosterone therapy increases the risk of prostate cancer or benign prostatic hyperplasia.
– There is also no evidence that testosterone treatment will convert subclinical prostate cancer to clinically detectable prostate cancer.
* Provocative new research evidence suggests that it is not high serum testosterone that is problematic for prostate cancer, but to the contrary that
it is low serum testosterone that is associated with worrisome cancer features and outcomes, such as high Gleason score, advanced stage of presentation, positive biopsy, and increased risk of biochemical recurrence after surgery.[13-15] Patients with prostate cancer and lower testosterone levels have undesirable prognosis factors and higher tumor burden before treatment onset.[15] These findings reinforce the idea that low testosterone levels pretreatment are related to a poor prognosis in prostate cancer.
* New experimental research has uncovered mechanisms that explain how low testosterone levels may be detrimental for prostate health, and support the view that testosterone therapy actually may have beneficial effects with regard to Prostate cancer. Specifically, testosterone promotes less aggressive prostate cancer types and inhibit dedifferentiation (i.e. metastasis [16]) in some prostate cancer cell lines.[17-19]
What is known – The Androgen Hypothesis
The idea that testosterone has detrimental effects on the prostate, the so called “androgen hypothesis” arose from two small studies in the 1940s in which men with metastatic prostate cancer demonstrated clinical and biochemical improvement with androgen deprivation via castration or estrogen treatment, and conversely rapid Prostate cancer progression with testosterone administration.[4, 5] Rather than concluding that exogenous testosterone attenuates the effects of surgical castration, the authors concluded that prostate cancer is activated by testosterone.[20] Notably, these observations were made in a special population (castrated men) and are therefore not relevant to testosterone therapy in hypogonadal men.[6]
Medical students and doctors have since been taught that high testosterone levels promote the development of prostate cancer, that low testosterone is protective, and that the administration of testosterone to a man with existing prostate cancer is like “pouring gasoline on a fire.”[3] This fear is also the most common reason for doctors’ reluctance to prescribe testosterone replacement therapy, even in hypogonadal men [1, 2] , which unnecessarily deprives many hypogonadal men of clinical benefits.
Although the dramatic effects of androgen deprivation therapy (ADT) in prostate cancer are indisputable [21], a large body of current evidence fails to support the concept that increasingly high levels of testosterone or DHT lead to ever-greater growth of benign or malignant prostate tissue (see below). It is critical to note that the androgen hypothesis was accepted prior to the discovery of the androgen receptor and PSA (prostate specific antigen), and before the availability of reliable serum testosterone assays. It should therefore not be surprising that some predictions of the androgen hypothesis would turn out to be false when submitted to rigorous scientific investigation.
Paradigm Shift – The Saturation Model It has been conclusively demonstrated that prostate cancer risk is unrelated to endogenous serum androgen concentrations [7], and several studies show no correlation between endogenous testosterone and PSA or prostate volume.[22, 23] Thus, men with higher endogenous testosterone are at no greater risk for prostate cancer than men with lower serum testosterone.
It has been conclusively demonstrated that prostate cancer risk is unrelated to endogenous serum androgen concentrations [7], and several studies show no correlation between endogenous testosterone and PSA or prostate volume.[22, 23] Thus, men with higher endogenous testosterone are at no greater risk for prostate cancer than men with lower serum testosterone.
The incidence of prostate cancer during long-term (up to 20 years) testosterone therapy has been demonstrated to be equivalent to that expected in the general population.[10] In healthy men, administration of supra-physiologic doses of testosterone (weekly injections of 500-600 mg of testosterone enanthate to healthy volunteers for up to 16 weeks) resulted in no increase in PSA nor prostate volume.[24, 25] In hypogonadal men treated with testosterone, levels of PSA typically rise up to levels of eugonadal men, but stay within the normal range.[26] This elevation in PSA and prostate volume commonly occurs during the initial 3-6 months after initiation of testosterone therapy, and then stabilize, even with continued testosterone therapy.[27-30]
In line with these findings, two meta-analyses of testosterone therapy intervention studies, which focused on analyzing potential adverse effects of testosterone therapy, did not find any significant differences in prostate outcomes between testosterone therapy vs. placebo treated men.[8, 9] Specifically, the most recent meta-analysis published in 2010 demonstrated no difference in the rates of prostate cancer, the need for prostate biopsy, international prostate symptom score (IPSS), increase in PSA, or total number of prostate-related adverse events when comparing the testosterone group with the placebo group.[9]
To explain these finding, the androgen hypothesis has been replaced by the saturation model.[31] The saturation model explains the paradoxical observations that prostate tissue is exquisitely sensitive to changes in testosterone levels at low concentrations, but becomes insensitive to changes in androgen concentrations at higher levels.[31] This response is consistent with the observation that testosterone exerts its prostatic effects primarily via binding to the androgen receptor, and that maximal testosterone- androgen receptor binding is achieved at testosterone levels well below the physiologic range.[31]
Changes in testosterone levels below the point of maximal testosterone – androgen receptor binding can elicit substantial changes in prostate cancer growth, as seen with castration, or with testosterone administration to castrated or hypogonadal men. In contrast, once maximal testosterone – androgen receptor binding is reached, further increasing testosterone levels results in little further effect. Thus, there is a threshold where increasing testosterone levels reach a limit (the saturation point) beyond which there is no further induction of androgen-driven changes in prostate tissue growth, see figure 1.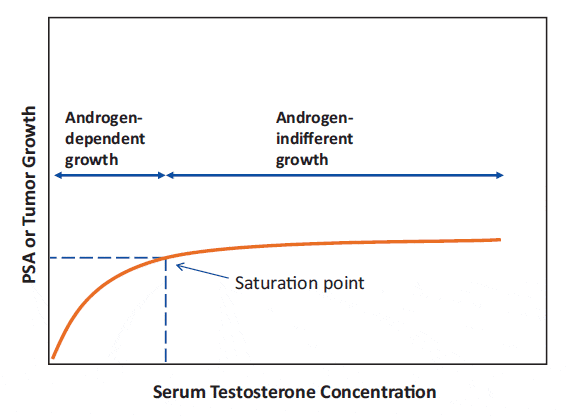
Figure 1: The saturation model. Increasing testosterone levels produce increasing prostate tissue growth, as reflected by PSA elevation, until a limit is reached (the saturation point), beyond which there is no further ability to induce androgen-driven changes.
From Khera et al. 2014
Maximal testosterone- androgen receptor binding (i.e., saturation) occurs at fairly low androgen concentrations. In clinical practice, the saturation point appears to be approximately 230 ng/dL (8 nmol/L). However, there is inter-individual variation in the saturation point. Other physiologic mechanisms may contribute, as well. For example, in aging men with low testosterone levels, 6 months of TRT normalized serum testosterone levels but had little effect on prostate tissue androgen levels, suggesting the presence of local regulatory mechanisms.[32] It should be noted that different tissues likely have different saturation points.
This explains why dramatic changes in PSA are noted when testosterone levels are treated into or out of the castration range, and when hypogonadal men get TRT treatment, whereas minimal or no PSA changes occur when higher doses are administered to most men. Thus, according to the saturation model, the initial modest PSA elevation and prostate growth within the reference range is a normal physiologic response to TRT in hypogonadal men. Therefore, testosterone-induced prostate growth and modest PSA elevations should not preclude hypogonadal men from testosterone substitution therapy.[26]
Summary
The long-held belief that prostate cancer risk is related to high testosterone levels (the androgen hypothesis) is not supported by clinical data. The saturation model and paradigm change that it brings to old inaccurate reasoning is that testosterone has a finite ability to stimulate prostate cancer growth.
The saturation model explains the paradoxical observations that prostate tissue is sensitive to changes in testosterone levels at low concentrations, but becomes insensitive to changes in testosterone levels at higher levels. Men with high testosterone levels are not at increased risk of developing prostate cancer, low testosterone levels provide no protection against the development of prostate cancer, and some men with untreated prostate cancer have received testosterone therapy without evidence of prostate cancer progression.[33]
Current evidence indicates that maximal testosterone-stimulated prostate cancer growth is achieved already at low sub-optimal testosterone levels. Provocative new research suggests that it is not high serum T that is problematic for prostate cancer, but to the contrary that it is low serum T that is associated with worrisome cancer features and outcomes, in that androgens promote less aggressive prostate cancer types and inhibit metastasis of established prostate cancer.
Therefore, no hypogonadal man should be deprived of testosterone therapy because of the unfounded old-school belief about the testosterone-prostate relationship. If you doctor still tells you that he won’t prescribe you testosterone therapy because “it will cause prostate cancer”, get another doctor!
References:
1. Gooren, L.J. and H.M. Behre, Diagnosing and treating testosterone deficiency in different parts of the world: changes between 2006 and 2010. Aging Male, 2012. 15(1): p. 22-7.
2. Atan, A., et al., Serum testosterone level, testosterone replacement treatment, and prostate cancer. Adv Urol, 2013. 2013: p. 275945.
3. Khera, M., et al., A new era of testosterone and prostate cancer: from physiology to clinical implications. Eur Urol, 2014. 65(1): p. 115-23.
4. Huggins, C. and C.V. Hodges, The effect of castration, of estrogen and of androgen injection on serum phosphatase in metastatic carcinoma of the prostate. Cancer Res 1941;1:293–7, 1941.
5. Huggins, C., R.E.J. Stevens, and C.V. Hodges, The effect of castration, of estrogen and of androgen injection on serum phosphatase in metastatic carcinoma of the prostate. Arch Surg 1941;43:209-223, 1941.
6. Morgentaler, A., Guilt by association: a historical perspective on Huggins, testosterone therapy, and prostate cancer. J Sex Med, 2008. 5(8): p. 1834-40.
7. Roddam, A.W., et al., Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. J Natl Cancer Inst, 2008. 100(3): p. 170-83.
8. Calof, O.M., et al., Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci, 2005. 60(11): p. 1451-7.
9. Fernandez-Balsells, M.M., et al., Clinical review 1: Adverse effects of testosterone therapy in adult men: a systematic review and meta-analysis. J Clin Endocrinol Metab, 2010. 95(6): p. 2560-75.
10. Feneley, M.R. and M. Carruthers, Is testosterone treatment good for the prostate? Study of safety during long-term treatment. J Sex Med, 2012. 9(8): p. 2138-49.
11. Raynaud, J.P., et al., Prostate-specific antigen (PSA) concentrations in hypogonadal men during 6 years of transdermal testosterone treatment. BJU Int, 2013. 111(6): p. 880-90.
12. Wang, C., et al., Investigation, treatment, and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA, and ASA recommendations. Eur Urol, 2009. 55(1): p. 121-30.
13. Morgentaler, A., Rapidly shifting concepts regarding androgens and prostate cancer. ScientificWorldJournal, 2009. 9: p. 685-90.
14. Garcia-Cruz, E., et al., Low testosterone level predicts prostate cancer in re-biopsy in patients with high grade prostatic intraepithelial neoplasia. BJU Int, 2012. 110(6 Pt B): p. E199-202.
15. Garcia-Cruz, E., et al., Low testosterone levels are related to poor prognosis factors in men with prostate cancer prior to treatment. BJU Int, 2012. 110(11 Pt B): p. E541-6.
16. Brawn, P.N. and V.O. Speights, The dedifferentiation of metastatic prostate carcinoma. Br J Cancer, 1989. 59(1): p. 85-8.
17. Hatzoglou, A., et al., Membrane androgen receptor activation induces apoptotic regression of human prostate cancer cells in vitro and in vivo. J Clin Endocrinol Metab, 2005. 90(2): p. 893-903.
18. Sonnenschein, C., et al., Negative controls of cell proliferation: human prostate cancer cells and androgens. Cancer Res, 1989. 49(13): p. 3474-81.
19. Chuu, C.P., et al., Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res, 2005. 65(6): p. 2082-4.
20. Kava, B.R., To treat or not to treat with testosterone replacement therapy: a contemporary review of management of late-onset hypogonadism and critical issues related to prostate cancer. Curr Urol Rep, 2014. 15(7): p. 422.
21. Schroder, F., et al., Androgen deprivation therapy: past, present and future. BJU Int, 2012. 109 Suppl 6: p. 1-12.
22. Monath, J.R., et al., Physiologic variations of serum testosterone within the normal range do not affect serum prostate-specific antigen. Urology, 1995. 46(1): p. 58-61.
23. Monda, J.M., et al., The correlation between serum prostate-specific antigen and prostate cancer is not influenced by the serum testosterone concentration. Urology, 1995. 46(1): p. 62-4.
24. Bhasin, S., et al., The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med, 1996. 335(1): p. 1-7.
25. Cooper, C.S., et al., Effect of exogenous testosterone on prostate volume, serum and semen prostate specific antigen levels in healthy young men. J Urol, 1998. 159(2): p. 441-3.
26. Behre, H.M., J. Bohmeyer, and E. Nieschlag, Prostate volume in testosterone-treated and untreated hypogonadal men in comparison to age-matched normal controls. Clin Endocrinol (Oxf), 1994. 40(3): p. 341-9.
27. Meikle, A.W., et al., Prostate size in hypogonadal men treated with a nonscrotal permeation-enhanced testosterone transdermal system. Urology, 1997. 49(2): p. 191-6.
28. Moon, D.G., et al., The efficacy and safety of testosterone undecanoate (Nebido((R))) in testosterone deficiency syndrome in Korean: a multicenter prospective study. J Sex Med, 2010. 7(6): p. 2253-60.
29. Snyder, P.J., et al., Effects of testosterone replacement in hypogonadal men. J Clin Endocrinol Metab, 2000. 85(8): p. 2670-7.
30. Wang, C., et al., Long-term testosterone gel (AndroGel) treatment maintains beneficial effects on sexual function and mood, lean and fat mass, and bone mineral density in hypogonadal men. J Clin Endocrinol Metab, 2004. 89(5): p. 2085-98.
31. Morgentaler, A. and A.M. Traish, Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur Urol, 2009. 55(2): p. 310-20.
32. Marks, L.S., et al., Effect of testosterone replacement therapy on prostate tissue in men with late-onset hypogonadism: a randomized controlled trial. JAMA, 2006. 296(19): p. 2351-61.
33. Morgentaler, A., et al., Testosterone therapy in men with untreated prostate cancer. J Urol, 2011. 185(4): p. 1256-60.






Hi Will, the article states: “This response is consistent with the observation that testosterone exerts its prostatic effects primarily via binding to the androgen receptor (AR), and that maximal testosterone-AR binding is achieved at testosterone levels well below the physiologic range.[8]” So, what about the evidence that supraphysiological levels of T elicits upregulation of ARs? (I left this comment in FB also).
AR upregulation in what tissue?
In muscle tissue. Is prostate tissue different?
Yes, different tissues have different dose-response curves and different saturation points.
Thank you Monica.
Monica, I very much enjoy your detailed and most relevant articles, especially this one. Since I have been prescribed T your article answered a major question that has been a concern. Thank you!
Bruce, I’m very glad to hear that. 🙂
Thanks Monica. I have been reading some of your research articles lately and am very impressed. This one agrees with other data I’ve read by several of the writers in Muscular Development. This is of great interest to me as I had prostate cancer in 2012 and 2013. My test levels prior treatment was 839ng. After a anti androgen shot, it was under 20ng in under two weeks. The surgery went great. I had 5 weeks of radiation and 160 radioactive seeds implanted in me. It’s been a while now, and my PSA has remained low. For several months now my doctor has tried everything to bring my test levels up. Now I’m getting test shots every other week. So far I’ve had 4 and the last one he upped the ante slightly. Should I speak to him about going larger? From what I just read, it sounds as if I will have a greater chance for it to increase my PSA at these lower levels. Is that the take home?
Eldon, the nitty-gritty issues in your CONTEXT are:
1. Is your PC (prostate cancer) cured. If it is, then the use of TRT (testosterone replacement therapy) will not induce new PC. Thus, TRT is a stress test for any residual PC.
2. After your treatment of PC, which appears to have involved an LH-RH agonist drug (not an anti-androgen shot as you state), and a radical prostatectomy and also radiation with external beam and seeds (sounds like there are many key points to your story not being mentioned), your T levels were low. This should have been explored (maybe it was) as to the cause. I would speculate that it might be due to the RT (radiation therapy) leading to injury to the gonads. If so, then simply obtaining an LH and FSH level would have revealed very elevated levels of both. Also, if I were treating you I would not be giving you TRT in the form of shots every two weeks. I would use a topical form of TRT that you could apply, which is a heck of a lot easier than going to a doctor’s office twice a month.
3. Other issues should be explored relating to your entire case. This forum is not the proper place for that.
Stephen B. Strum, MD, FACP
Medical Oncologist Specializing in PC since 1983